美國 HHS 啟動 Operation TrialBlazer:臨床試驗規則要變了,新藥競爭也要變了
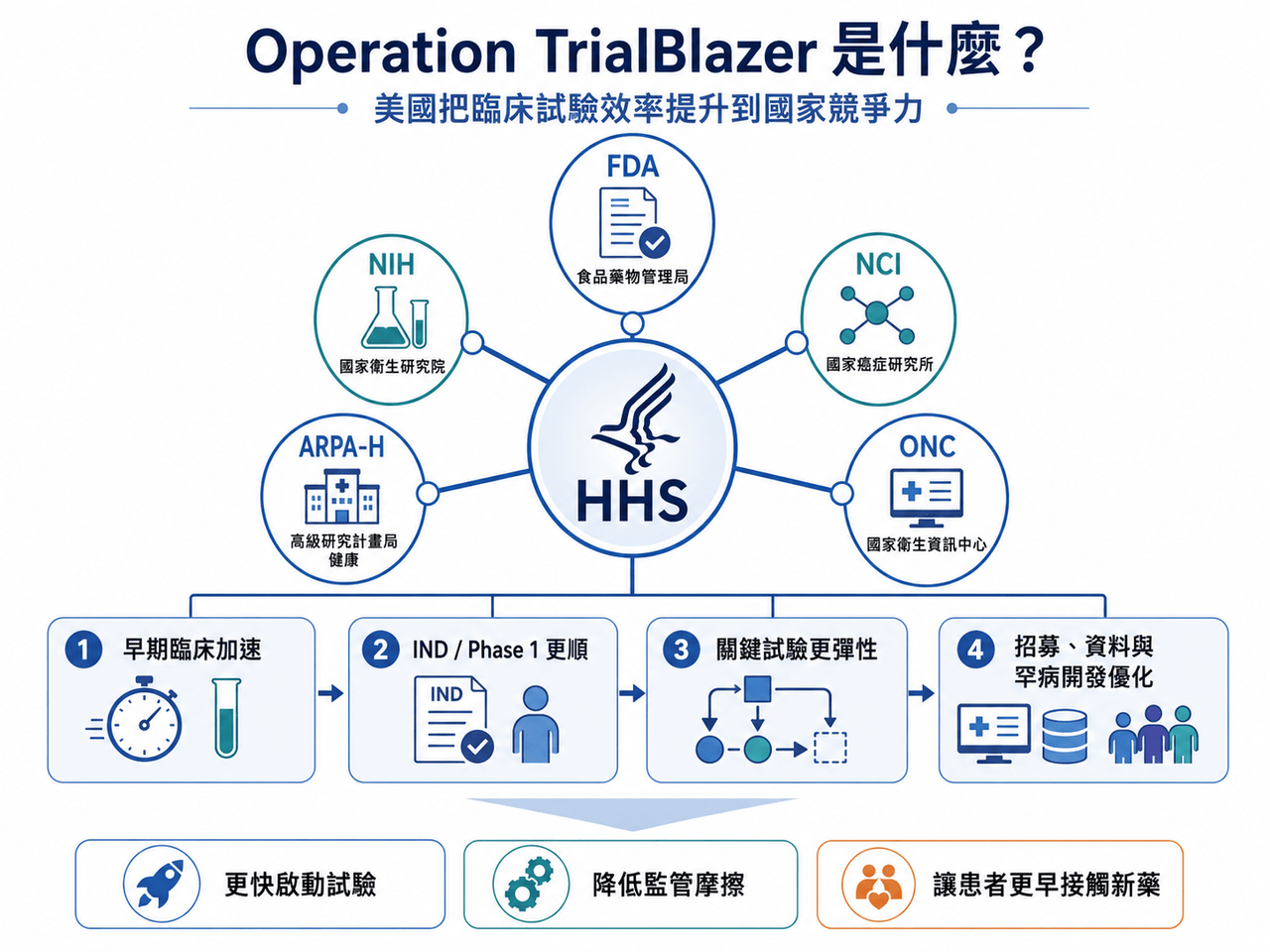
2026 年 6 月 22 日,美國衛生與公共服務部,也就是 HHS,啟動了一項名為 Operation TrialBlazer 的部門級臨床試驗改革計畫。
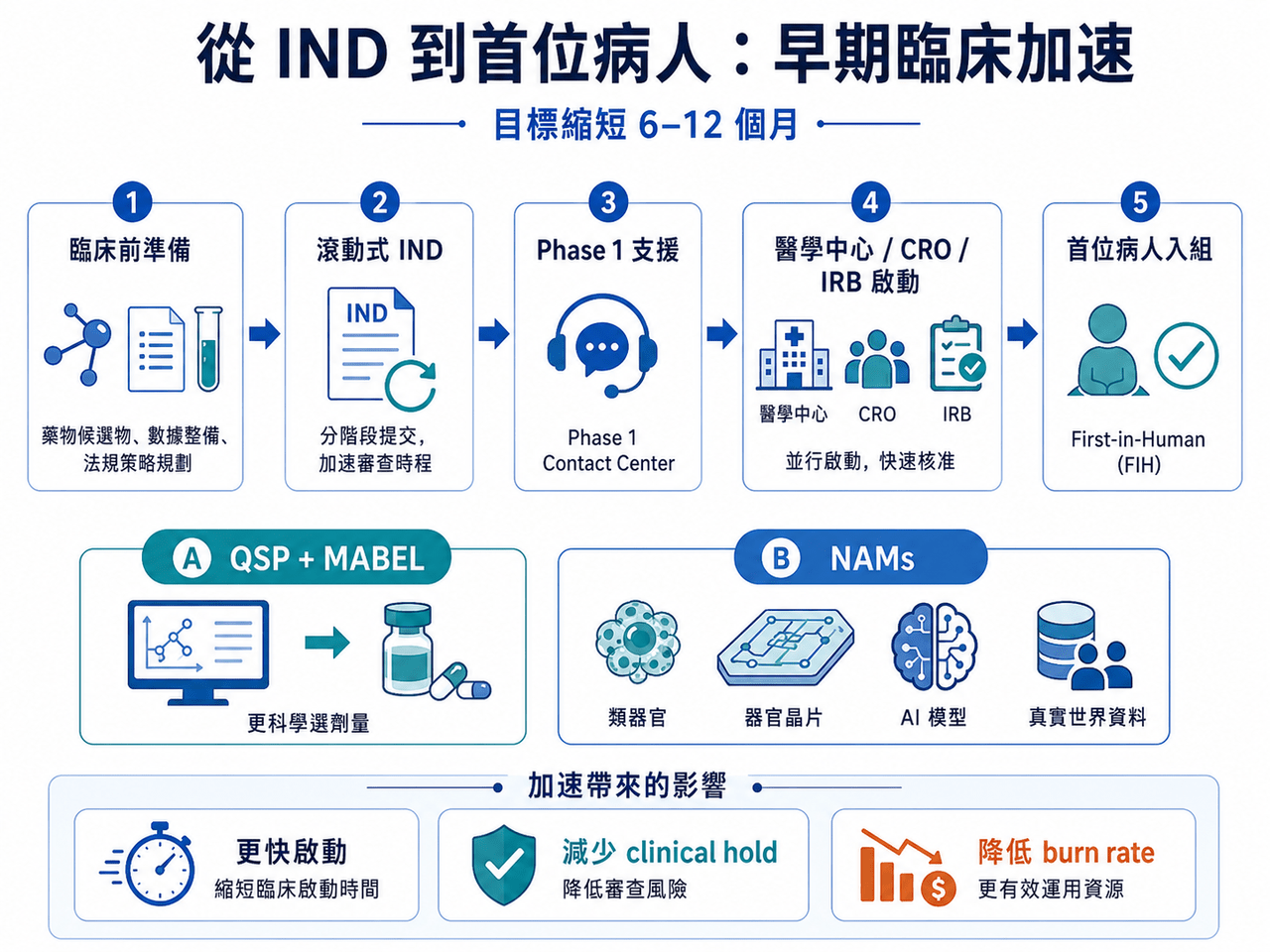
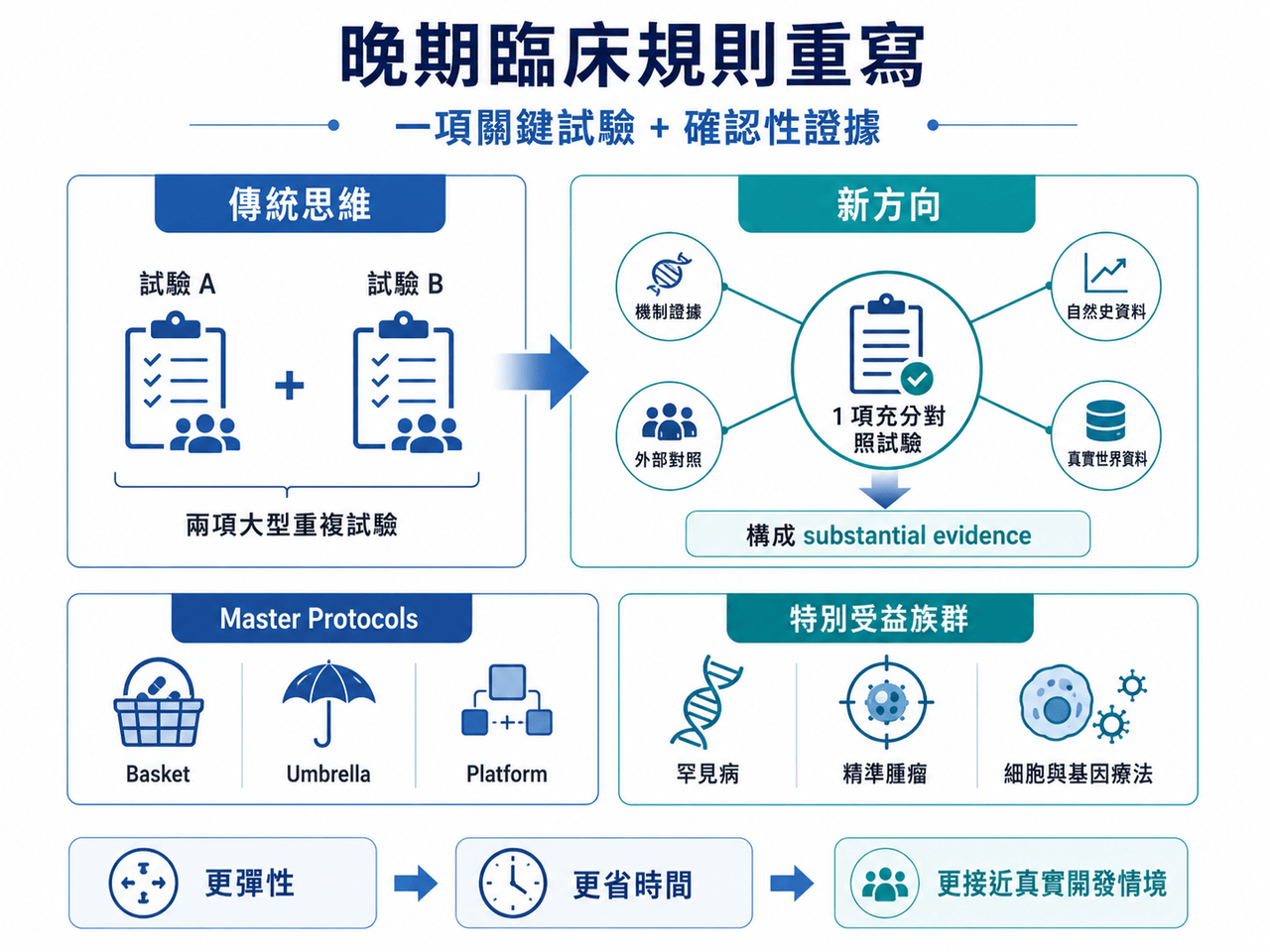
這不是 FDA 單一部門的小修小補,而是把 FDA、NIH、NCI、ARPA-H、ONC 等核心機構一起拉進來,試圖從早期臨床、IND 申報、劑量選擇、關鍵性試驗設計、電子病歷招募、罕見病開發,到受試者報酬合規,重新調整美國臨床試驗體系。
HHS 官方聲明把目標講得很直白:恢復美國在臨床研究的領先地位,加快救命療法開發,讓患者更快接觸創新藥。
HHS 部長 Robert F. Kennedy Jr. 也在聲明中表示,美國曾經領跑全球醫療創新,現在要重新做到這一點。
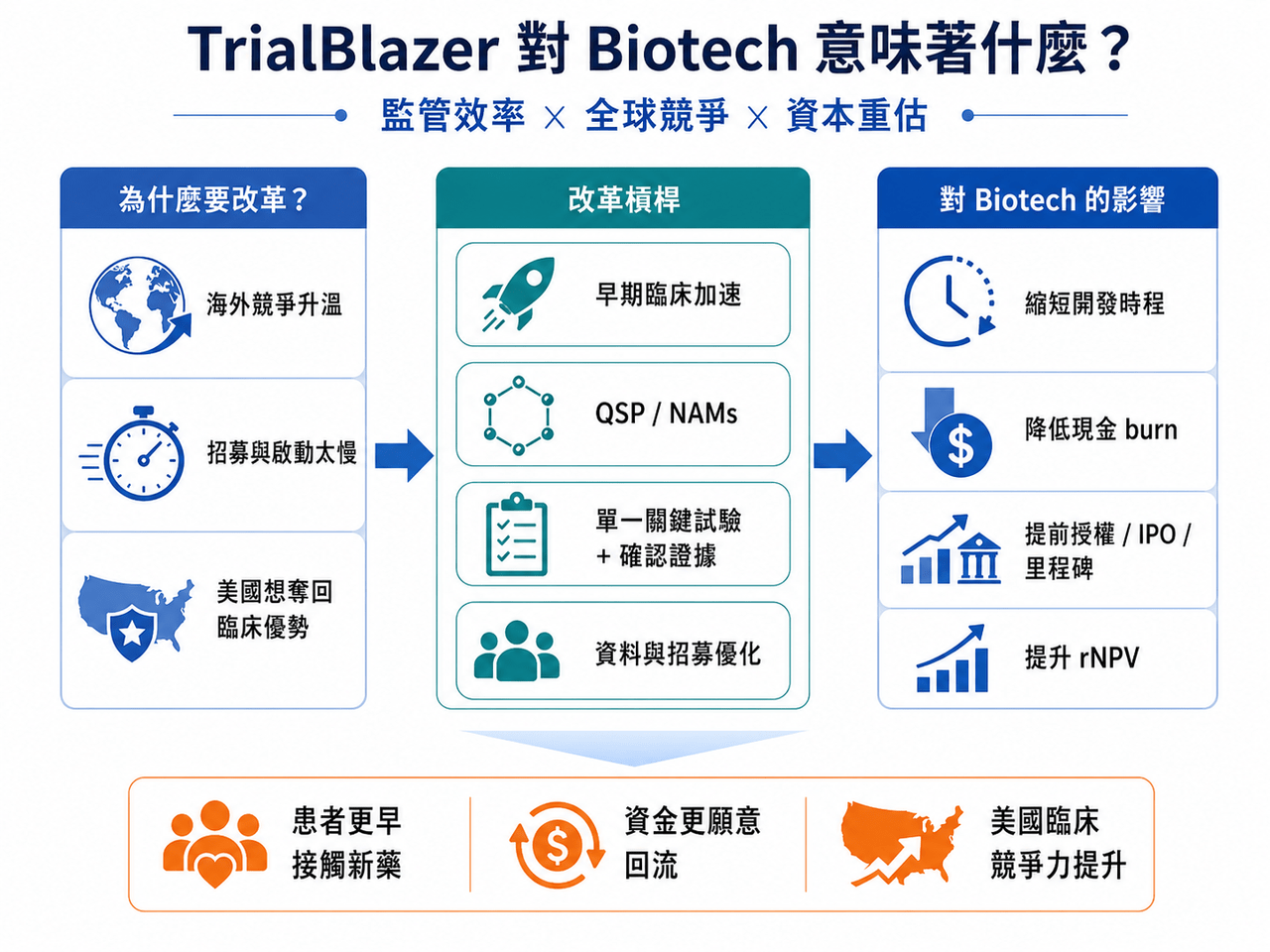
📌 這句話背後的意思其實很清楚:美國覺得自己慢了。
尤其是在全球臨床試驗競爭愈來愈激烈、中國臨床開發效率快速提升、Biotech 資本又重新尋找高彈性資產的背景下,FDA 和 HHS 不能再用舊節奏面對新產業。
這次改革真正重要的,不只是幾條技術性指引,而是它釋放出一個訊號:
📌 美國正在把臨床試驗效率,提升到國家競爭力層級。